Методы определения тяжелых металлов в сырье и готовой пищевой продукции.
Пищевые продукты и напитки имеют сложный химический состав, при этом тяжелые металлы могут присутствовать в очень малых количествах, поэтому необходимо выбирать методы анализа с низким пределом обнаружения и высокой селективностью. Для определения тяжелых металлов в растительном сырье и готовых пищевых продуктах используют различные методы анализа, среди них спектральные методы анализа (атомно-абсорбционный, атомно-эмиссионный, спектрофотометрический и фотометрический анализ), электрохимические (инверсионная вольтамперометрия, полярография), рентгенофлуоресцентный анализ и т.д.
Для определения содержаний элементов Ca, Fe, Ti, Mn, Cr, Cu, Ni, Zn, Br, Sr, Ba и Pb в лекарственном растении красноднев малый применили метод рентгенофлуоресцентного анализа (РФА), который позволяет анализировать образцы без разрушения их основы химическим и термическим воздействием. Авторами работы [20] приведены пределы обнаружения методики ренгенофлуоресцентного определения элементов в растениях.
Для определения свинца в томатном, апельсиновом, ананасовом соках методом переменно-токовой полярографии используют кислотное озоление пробы с последующей обработкой пероксидом водорода. Определению мешает Sn (IV) и значительный избыток меди. Методика определения свинца в виноградном соке основана на экстракционном концентрировании 8-оксихинолината свинца бутанолом-1 при pH 10-12 и последующем полярографировании экстракта на фоне 0,75 М раствора LiCl+1M HCl в смеси (1:4) этанола и воды. Разработанная методика полярографического определения свинца без минерализации пробы пригодна для определения до 0,01 м к г/мл свинца. Определение свинца в апельсиновом соке методом дифференциальной импульсной полярографии проводится на фоне 0,1М СН3СООН + 0,5М СН3СООNa (pH 4,5) в присутствии тритона Х-100. Предел обнаружения 0,01 мкг/л [21].
Изучена возможность определения Pb, Cd и Cu в образцах меда без предварительного разложения пробы с использованием анодной дифференциальной импульсной инверсионной вольтамперометрии в проточной ячейке. Показано, что вольтамперометрическое определение Cd2+ и Pb2+ в водном растворе меда возможно при использовании индикаторного ртутного электрода типа "висячая капля" после модифицирования его Тритоном Х-100. Предел обнаружения по Pb2+, Cd2+ 20 мкг/л. Определение Cd2+, Cu2+ и Pb2+ возможно при использовании пленочного ртутного электрода на стеклоуглеродной подложке [22].
Наиболее часто для определения тяжелых металлов в пищевых продуктах применяют спектральные методы анализа. Метод твердофазной спектрофотометрии (ТФС) эффективен для определения Cu, Pb, Zn, Fe (III), Cd, Hg, Sn(IV) в пищевых продуктах. Твердофазная спектрофотометрия позволяет проводить фотометрическое определение в фазе сорбента. Изучено комплексообразование ряда токсичных элементов с красителями кислотного и основного типа в твердой фазе [23].
Атомно-эмиссионная спектрометрия с индуктивно связанной плазмой (АЭС-ИСП) характеризуется низкими значениями нижних границ интервалов определяемых содержаний элементов в растворах, широким диапазоном интервала определяемых содержаний элементов в растворах, широким диапазоном интервала линейности градуировочных характеристик, высокой сходимостью результатов единичных определений, низким уровнем влияний сопутствующих компонентов на результаты анализа, информативностью, простым решением проблемы
приготовления адекватных составу анализируемого раствора образцов сравнения [24].
Методика анализа, включающая автоклавную пробоподготовку в сочетании с АЭС-ИСП, применена для определения Cu, Zn, Mn, и Cr в плодах и цветках разных видов боярышника. Уровни содержаний сопутствующих элементов не мешают определению Cu, Zn, Mn, и Cr в интервале содержаний 10-3 – 10 -6 масс.% [24].
Метод атомно-абсорбционной спектрометрии (ААС) обладает высокой экспрессностью и точностью, низким пределом обнаружения . Его основное преимущество перед другими методами в высокой селективности, простоте подготовки проб к анализу и возможности определения нескольких элементов из одного раствора по единой методике [6].
Был предложен простой метод для прямого определения меди в различных образцах чая пламенной атомно-абсорбционной спектрометрией (ПААС). Метод ПААС был применен для определения меди в пяти различных образцах чая. Предел обнаружения 0,05 мг/мл. Исследования показали, что определению меди методом ПААС не мешают высокие концентрации других элементов [25].
Электротермическая атомно-абсорбционная спектрометрия (ЭТААС) используется для массового определения низких уровней концентрации металлов в различных типах образцов, позволяет напрямую анализировать твердые образцы. Высокая чувствительность метода позволяет исключить стадии экстракции и предварительного концентрирования. Однако по сравнению с ПААС он менее экспрессен и при анализе в графитовой печи в отличие от пламенных атомизаторов обязательно требуется коррекция фона. Недостатком можно считать и то, что ЭТААС не используется для анализа тугоплавких элементов [26].
Был предложен прямой атомно-абсорбционный анализ твердых пищевых продуктов, основанный на использовании электротермического атомизатора, в котором зоны испарения и атомизации разделены графитовыми фильтрами. В таком атомизаторе твердую пробу дозируют в испаритель, отделенный от аналитической зоны перегородкой - фильтром из пористого графита. При нагреве испарителя пары пробы поступают в аналитическую зону после фильтрации через графитовый фильтр, что исключает выброс неатомизированных частичек. Способ прямого экспрессного атомно-абсорбционного определения кадмия в твердых пробах с помощью модифицированной кюветы Львова с графитовым фильтром-перегородкой позволяет устранить стадию перевода твердой пробы в раствор, подавить неселективное поглощение, существенно сократить время, необходимое для проведения анализа. Предел определения кадмия при массе порошковой пробы 10 мг составляет 4,6*10-3% [27].
Существует ГОСТ 30178-96 «Сырье и продукты пищевые. Атомно-абсорбционный метод определения токсичных элементов», который устанавливает метод определения свинца, кадмия, меди, цинка и железа в пищевом сырье и продуктах. Метод основан на минерализации продукта способом сухого или мокрого озоления и определении концентрации элемента в растворе минерализата методом пламенной атомной абсорбции [28].
ГОСТ 26932-86 «Сырье и продукты пищевые. Методы определения свинца» и ГОСТ 26933-89 «Сырье и продукты пищевые. Методы определения кадмия» для определения свинца и кадмия в поваренной соли предлагают атомно-абсорбционный и полярографический методы [29, 30]. Атомно-абсорбционный метод основан на предварительном концентрировании свинца и кадмия с последующим их определением на атомно-абсорбционном спектрофотометре в пламени ацетилен-воздух. Полярографический метод основан на сухой минерализации (озолении) пробы с использованием в качестве вспомогательного средства азотной кислоты и количественном определении свинца и кадмия полярографированием в режиме переменного тока [29,30].
Таким образом, для определения тяжелых металлов в пищевых продуктах наиболее широко используются спектральные методы анализа, в частности атомно-абсорбционный и атомно-эмиссионный, так как они достаточно селективны, экспрессны, имеют низкие пределы обнаружения и высокую чувствительность.
Методы определения ртути в пищевых продуктах
Содержание ртути в пищевых и других биологических объектах очень мало. Для определения ртути используют разные методы, среди них: колориметрический, атомно-абсорбционная спектрометрия (ААС), метод нейтронно-активационного анализа (НАА), электрохимические методы и т.д.
Колориметрический метод основан на переводе металла, содержащегося в навеске, в комплекс с дитизоном, который экстрагируют органическим растворителем и затем колориметрируют. Эти операции длительны; предел обнаружения составляет около 0,05 мг/кг. Для определения требуется большая навеска (5 г) образца. Вследствие маленького предела обнаружения этот метод нельзя использовать для анализа биологических объектов [19].
При нейтронно-активационном анализе содержание ртути определяют измерением излучения изотопов 203Hg и 197mHg. Гамма спектр изотопа 203Hg показан на рис.5. При химическом выделении изотопов ртути из облученного материала радиоактивность препаратов измеряют на простых пересчетных установках. Содержание ртути определяют относительным методом, т.е. путем сравнения активности пробы и эталона. Чувствительность определения ртути при облучении потоком 1010 нейтрон/см2•сек при весе пробы 1 г составляет 10-5-10-6 %, при потоках 1013-1014 нейтрон/см•сек можно определять содержание ртути 10-7-10-8%[23].
![Рис.5. Гамма спектр изотопа 203Hg[23].](img\ris5.jpg)
К недостаткам метода можно отнести: малую доступность источников активирующих частиц, необходимость защиты от радиоизлучений и высокую стоимость оборудования.
При анализе различных объектов на валовое содержание ртути возникает ряд проблем как общих для всех методов анализа, так и специфических для инверсионной вольтамперометрии (ИВА) [24]. Как правило, не все соединения ртути могут давать аналитический сигнал, поэтому необходим перевод их в аналитическую форму. Для определения следов ртути в водах методом ИВА широко применяют золотой и золото-графитовый дисковые электроды. Величина тока пика ртути (II) зависит от количества золота на поверхности электрода. Оптимальной оказалась концентрация золота (III) 4•10-6 М. При большей концентрации золота пик ртути выше, но требуется и большее время для электрохимической очистки электрода, иначе пики растут от опыта к опыту. На аналитический сигнал ртути заметно влияют хлорид-ионы. С увеличением их концентрации пик увеличивается и смещается к более отрицательным значениям потенциалов, что указывает на образование устойчивых комплексов ртути с хлорид-ионами вероятностного состава HgCl+.
Инверсионная дифференциальная импульсная вольтамперометрия позволяет определять ртуть (II) при концентрациях ниже 10-9 М в оптимальных условиях протекания электродного процесса, например на золотом индикаторном электроде [25]. Методика находит ограниченное применение, так как требуется сложное оборудование и специальная предварительная подготовка электрода. На углеродном электроде, без использования вспомогательных элементов, нижняя определяемая концентрация ионов ртути (II) равна 4•10-6-4•10-9 М. Если поверхность углерода предварительно покрывают тонкой золотой пленкой, она снижается до 5•10-10 при сmin, равной 5•10-11 М, на дисковом вращающемся электроде, в условиях повышенного массопереноса на стадии электронакопления. Снижение сmin при нанесении пленки золота объясняется как облегчением осаждения ртути за счет улучшения смачиваемости электрода ртутью путем амальгамирования, так и образованием интерметаллического соединения ртути и золота.
Более перспективна из практических соображений методика инверсионного определения ионов ртути (II) на импрегнированном графитовом электроде в присутствии ионов золота (II), которые используются для консервации водных растворов при определении низких концентраций ионов ртути (II). Образовавшийся «in situ» золото-графитовый электрод пригоден для определения ионов ртути (II) в диапазоне концентраций 10-9-2•10-6 М без сложной предварительной подготовки. Хотя импрегнированный графитовый электрод обладает хорошими электроаналитическими свойствами, последние могут варьироваться в широком диапазоне в зависимости от способа его изготовления.
Эти недостатки можно устранить при использовании цилиндрического микроэлектрода из углеродного волокна, который характеризуется улучшенными аналитическими свойствами, из-за его микронных размеров и хорошей воспроизводимости. В инверсионной дифференциальной импульсной вольтамперометрии в качестве аналитического сигнала, функционально связанного с концентрацией ртути (II) в растворе, можно использовать как величину тока пика ионизации ртути In, так и площадь под пиком Qn [25]. Данным методом можно анализировать воды различного происхождения, так как достигнутая экспериментально минимальная определяемая концентрация ртути гораздо ниже 1•10-8 М.
В статье «Высокочувствительные ионоселективные электроды на основе дитиакраун-эфиров для определения ртути в образцах рыб» исследованы электрохимические свойства мембран на основе макроциклических соединений, содержащих в полиэфирном кольце два атома серы [26].Электроды с высокой чувствительностью и селективностью к ртути (II) можно создать, вводя в мембраны тиакраун-эфиры с высокой липофильностью и макроциклической полостью, соответствующей ионному радиусу определяемого катиона, например на основе новых дитиакраун-эфиров (I) и (II):
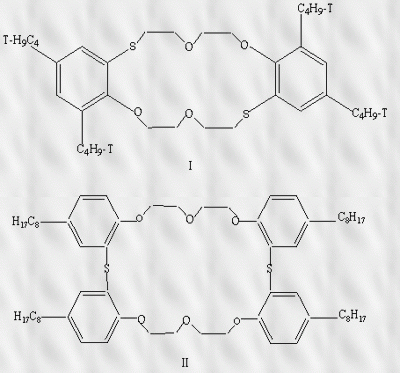
При несоответствии ионного радиуса ртути (II) размерам полости дитиакраун-эфира электродные свойства Hg2+-селективного электрода ухудшаются. Так, ионселективный электрод, основанный на тиакраун-эфире (I), по чувствительности на 2 порядка уступает Hg2+-селективному электроду на основе тиакраун-эфира (II). Показано, что ионселективный электрод на основе вещества (II) обладает химической устойчивостью и воспроизводимостью характеристик этого сенсора в растворах сложного состава.
При организации автоматического аналитического контроля за содержанием в питьевых, поверхностных и сточных водах таких нормируемых компонентов, как ртуть, целесообразно использовать простые методы и методики с минимальным набором доступных реагентов, например, проточно-инжекционный анализ (ПИА) с ионометрическим окончанием [27]. Но так как катионы ртути существуют в растворах преимущественно в форме комплексных частиц, прямое потенциометрическое определение с использованием ионселективных электродов (ИСЭ) возможно лишь в редких случаях.
Установка для ПИА представлена на рис.6.
![Рис.6. Принципиальная схема установки для проточно-инжекционного анализа. 1,2-перистальтические насосы, 3-клапан, 4-рН-метр, 5-КСП-4, 6-проточная потенциометрическая ячейка, 7-сорбционная колонка, 8,9-переключатели потока, 10-реакционная спираль [28].](img\ris7.jpg)
Лучшими динамическими и аналитическими характеристиками обладает жидкостной электрод с мембраной на основе нитробензольного раствора ионного ассоциата кристаллического фиолетового и иодидного комплекса ртути. Большое значение для оптимальной работы проточно-инжекционной системы имеет выбор потока-носителя, так как он оказывает влияние на стабильность базовой линии, чувствительность и производительность анализа. Из нескольких исследованных вариантов лучшим оказался 0,1 М раствор KI. При непосредственном введении пробы в систему ПИА в оптимальных условиях предел обнаружения составляет 1•10-6 моль/л. Этого недостаточно для определения ртути в большинстве объектов окружающей среды. С целью снижения предела обнаружения в схему анализа водных растворов включают этап концентрирования (сорбционное концентрирование ртути (II) проводят в ионообменной колонке с использованием ионообменника в калиевой форме с размерами частиц 0,25-0,5 мм). Минимально определяемая концентрация ртути данным методом является 1•10-8 М [27].
Обращено-фазовая ВЭЖХ с доколоночным получением производных является перспективным методом разделения и одновременного определения следов ионов металлов [28]. В настоящее время разработана новая методика одновременного определения ионов свинца, кадмия и ртути в виде хелатов с тетра-(4-хлорфенил)-порфирином (Т4ХФП) с использованием в режиме реального времени концентрирования и обращено-фазовой высокоэффективной жидкостной хроматографии. Хелаты Hg- Т4ХФП, Pb- Т4ХФП и Cd- Т4ХФП вводят в дозатор и с помощью подвижной фазы направляют в концентрирующую колонку. Хелаты адсорбируются в верхней части концентрирующей колонки. Путем переключения шестиходового крана подается обратный поток подвижной фазы, и удержанные хелаты вымываются в аналитическую колонку. Разделяют в градиентном режиме. Диапазон линейности градуировочного графика 0,01-120 мкг/л для всех хелатов. Пределы обнаружения для Pb, Cd и Hg равны соответственно 2.0, 1.5 и 2.0 нг/л.
Атомно-абсорбционный спектрометрия (ААС) еще в начале 70-х годов практически вытеснила колориметрические методы при определении ртути в воде (питьевой, природной, сточной) и пищевых продуктах [29]. В настоящее время для определения ртути в основном используют ААС и атомно-флуоресцентную спектрометрию (АФС), причем атомизацию проб проводят методом холодного пара (ХП). Метод ААС ХП включает в себя окислительное разрушение органических соединений в пробе (этап пробоподготовки) и последующее восстановление катионной формы ртути раствором дихлорида олова (SnCl2) или натрия боргидрата (NaBH4). Образовавшаяся атомарная ртуть выделяется из раствора и потоком газа переносится или прямо в аналитическую кювету (этап измерения), или предварительно в блок накопления (обычно содержащий золотой сорбент) [29].
Ранее в методе АФC для атомизации пробы использовали пламя [30]. Предел обнаружения ртути в водном растворе составлял 2 p.p.b. В последствие в данном методе использовали электротермическую атомизацию или ХП. Интенсивность флуоресценции в воздухе снижена из-за гашения кислорода и азота. Замена воздуха аргоном давала более высокую чувствительность (в 86 раз). Это достигалось использованием амальгам с золотом или фазовым разделением с пористой трубкой политетрафлуороэтиленом. Метод АФС позволяет определять ртуть в воздухе и водных растворах в количестве порядка пикограмма.
К недостаткам ААС ХП и АФС ХП следует отнести необходимость проведения многостадийной и, зачастую, трудоемкой процедуры пробоподготовки для обеспечения полноты выхода ртути. Недостатком также является необходимость борьбы с эффектом «памяти» в реакционном сосуде, коммуникациях и в аналитической кювете [29].
Также существует метод определения метилртути и контроля общей ртути в морепродуктах с использованием ВЭЖХ и масс-спектрометрии с индуктивно-связанной плазмой [31]. Вытяжки из морепродуктов разделяют с помощью обращено-фазовой ВЭЖХ, где подвижной фазой является L-цистеин (т.к. в рыбе метилртуть связана в комплекс с цистеином или соединением подобным цистеину по составу), используя колонку С-18 (при комнатной температуре) и детектируют с помощью масс-спектрометрии с индуктивно связанной плазмой. Общую ртуть вычисляют суммированием метилртути и неорганической ртути, определенных в вытяжках. Для морепродуктов, содержащих 0,055-2,78 мг/кг метилртути и 0,014-0,137 мг/кг неорганической ртути, точность анализа - 0,5 % (относительное стандартное отклонение) для метилртути и для неорганической ртути - 9%. Извлечение суммарного аналита 94% для метилртути и 98% для неорганической ртути. Результаты по метилртути и неорганической ртути в эталоне согласованы с аттестованными значениями. Предел обнаружения 0,007 мг/кг метилртути и 0,005 мг/кг неорганической ртути в морепродуктах. Общие результаты по ртути, определенные этим методом, были эквивалентны результатам, определенным независимым методом (ААС методом холодного пара). Отношение метилртути к общей ртути, определенное этим методом, составляло 93-98% для рыбы и 38-48% для моллюсков.
В настоящее время для определения содержания общей ртути в пищевых продуктах действует ГОСТ 26927-86, основанный на колориметрическом определении ртути с дитизоном [32]. Этот метод характеризуется недостаточной воспроизводимостью, трудоемкостью и может рассматриваться как полуколичественный. Более точный атомно-абсорбцоинный метод определения ртути предусмотрен в указанном ГОСТе только для рыбных продуктов, кроме того, этот метод предусматривает использование зарубежного анализатора ртути типа «MAS-50».
Методические указания (МУ) 5178 являются модификацией атомно-абсорбцоинного метода определения ртути для различных пищевых продуктов с использованием отечественного анализатора типа «Юлия-2». Предлагаемый метод основан на мокром кислотном озолении пробы с последующим восстановлением ртути до металлической формы и количественном определении методом беспламенной атомной абсорбции на отечественном анализаторе типа «Юлия-2»
Отбор и подготовку проб к испытанию проводят в соответствии с нормативно-технической документацией на продукт или пищевое сырье [33].
Наверх